Abstract
In the current study, we examined the effects of the nonpsychoactive cannabinoid, cannabidiol, on the induction of apoptosis in leukemia cells. Exposure of leukemia cells to cannabidiol led to cannabinoid receptor 2 (CB2)-mediated reduction in cell viability and induction in apoptosis. Furthermore, cannabidiol treatment led to a significant decrease in tumor burden and an increase in apoptotic tumors in vivo. From a mechanistic standpoint, cannabidiol exposure resulted in activation of caspase-8, caspase-9, and caspase-3, cleavage of poly(ADP-ribose) polymerase, and a decrease in full-length Bid, suggesting possible cross-talk between the intrinsic and extrinsic apoptotic pathways. The role of the mitochondria was further suggested as exposure to cannabidiol led to loss of mitochondrial membrane potential and release of cytochrome c. It is noteworthy that cannabidiol exposure led to an increase in reactive oxygen species (ROS) production as well as an increase in the expression of the NAD(P)H oxidases Nox4 and p22phox. Furthermore, cannabidiol-induced apoptosis and reactive oxygen species (ROS) levels could be blocked by treatment with the ROS scavengers or the NAD(P)H oxidase inhibitors. Finally, cannabidiol exposure led to a decrease in the levels of p-p38 mitogen-activated protein kinase, which could be blocked by treatment with a CB2-selective antagonist or ROS scavenger. Together, the results from this study reveal that cannabidiol, acting through CB2 and regulation of Nox4 and p22phox expression, may be a novel and highly selective treatment for leukemia.
Marijuana has been suggested as a potent therapeutic agent alleviating such complications as intraocular pressure in glaucoma and cachexia, nausea, and pain in AIDS and cancer patients. A number of recent studies now suggest the possible use of these compounds for the treatment of cannabinoid receptor-expressing tumors. For example, anandamide was shown to inhibit the proliferation of the human breast cancer cell lines MCF-7 and EFM-19 in vitro (De Petrocellis et al., 1998). In addition, THC was shown to induce apoptosis in human prostate PC-3 cells and in C6 glioma cells in culture (Sanchez et al., 1998; Ruiz et al., 1999; Galve-Roperh et al., 2000). THC-induced apoptosis involved cannabinoid receptor-dependent (Sanchez et al., 1998; Galve-Roperh et al., 2000) or -independent pathways (Ruiz et al., 1999). Such studies have triggered interest in targeting cannabinoid receptors in vivo to induce apoptosis in transformed cells. To this end, cannabinoids were shown to inhibit the growth of C6 glioma cells in vivo (Sanchez et al., 2001). Furthermore, recent studies from our laboratory demonstrated that targeting cannabinoid receptors may be a novel approach to treating lymphoblastic disease (McKallip et al., 2002).
A significant limitation to the use of a number of these compounds is their unwanted psychotropic activity. Cannabidiol (CBD) is a nonpsychoactive derivative of marijuana that is currently being examined for its use in the treatment of cancer. For example, Massi et al. (2004) demonstrated that cannabidiol was capable of suppressing the proliferation of human glioma cell lines. In addition, the HL-60 myeloblastic cell line was shown to be sensitive to CBD-induced apoptosis, whereas monocytes from healthy subjects were relatively resistant to CBD-induced apoptosis, suggesting that CBD may be effective at treating acute myelogenous leukemia (Gallily et al., 2003).
Although a number of reports demonstrate the ability of CBD to induce apoptosis in tumor cells, little work has been done demonstrating CBD mechanism of action. Massi et al. (2004) found that apoptosis in human glioma cell lines after exposure to CBD was mediated through CB2 receptor and the generation of reactive oxygen species (ROS). The generation of ROS can play an important role in the induction of apoptosis in T cells undergoing either activation-induced cell death or activated T cell autonomous cell death (Hildeman et al., 2003). Furthermore, the regulation of ROS generation can be significantly affected by NAD(P)H oxidases (Suzuki et al., 1998; Lee et al., 2000). Numerous studies have been carried out examining the ability of compounds to induce apoptosis in tumor cells by increasing ROS production (Kang et al., 2004; Kim et al., 2004; Chang et al., 2005; Hu and Brindle, 2005; Lebedeva et al., 2005). However, little is known about the ability of cannabinoids or signaling through cannabinoid receptors to regulate the expression or activity of NAD(P)H oxidases and/or to control of ROS generation in leukemia. However in a recent study, the NAD(P)H oxidase Nox5, which plays a significant role in mediating Ca2+-dependent ROS generation, was shown to be expressed in lymph nodes and the spleen, suggesting a possible role of NAD(P)H oxidases in the regulation of ROS production in cells of the immune system (Banfi et al., 2001).
The observation that CBD can mediate apoptosis through cannabinoid receptor 2 (CB2) combined with results from our previous study, where we demonstrated that, compared with a human glioma cell line, a number of human leukemias and lymphomas expressed significantly higher levels of CB2, suggests the possibility that human leukemias and lymphomas may be highly sensitive to the CB2-mediated effects of cannabidiol (McKallip et al., 2002; Massi et al., 2004). Therefore, in the current study, we examined the potential use of cannabidiol in the treatment of lymphoblastic disease. Using the murine EL-4 leukemia and the human Jurkat and Molt-4 leukemia cell lines, we demonstrated that exposure to cannabidiol led to tumor killing by induction of apoptosis. Examination of the mechanism of cannabidiol-induced apoptosis revealed that cannabidiol was acting through CB2. Furthermore, we demonstrated the involvement of the NAD(P)H oxidases p22phox and Nox4 and the subsequent generation of reactive oxygen species in CBD-induced apoptosis. Together, these studies suggest that cannabidiol, acting through CB2, may be a novel regulator of NAD(P)H oxidase expression and that CBD may prove to be a potent nonpsychoactive and specific treatment of leukemia.
Materials and Methods
Mice. Adult female (6-8 weeks of age) C57BL/6 mice were purchased from the National Institutes of Health (NIH). The mice were housed in polyethylene cages and given rodent chow and water ad libitum. Mice were housed in rooms maintaining a temperature of 74 ± 2°F and a 12-h light/dark cycle.
Reagents. Cannabidiol initially dissolved in EtOH was obtained from Tocris Cookson Inc. (Ellisville, MO). Cannabidiol was further diluted with tissue culture medium for in vitro studies and PBS for in vivo studies. SR141716A [cannabinoid receptor (CB) 1-selective antagonist] and SR144528 (CB2-selective antagonist), were obtained from Sanofi Recherche (Montpellier, France). α-Tocopherol, diphenylene iodinium (DPI), capsazepine [CPZ; a vanilloid receptor 1 (VR1)-selective antagonist], apocynin, and N-acetylcysteine (NAC) were obtained from Sigma-Aldrich (St. Louis, MO). Antibodies specific for caspase-2, -3, -8, -9, -10, Bid, cytochrome c, and poly(ADP-ribose) polymerase were purchased from Cell Signaling Technology (Danvers, MA). Antibodies specific for p22pnox and Nox4 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Cell Lines. The murine lymphoma cell line EL-4 and the human leukemia cell lines Jurkat and MOLT-4 were maintained in RPMI 1640 medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal calf serum, 10 mM HEPES, 1 mM glutamine, 40 μg/ml gentamicin sulfate, and 50 μM 2-mercaptoethanol. Serum-free medium was used in assays examining the effect of cannabidiol on tumor cell viability and apoptosis.
Measurement of the Effect of Cannabidiol on Tumor Cell Viability in Vitro. Tumor cells were adjusted to 0.5 × 106 cells/ml in serum-free medium. Next, the cells (0.5 × 106) were cultured for 24 h in 24-well plates in 2 ml of serum-free medium in the presence or absence of various concentrations of cannabidiol. Finally, the cells were harvested and washed twice in PBS, and the viable cell count was determined by trypan blue dye exclusion.
Detection of Cannabidiol-Induced Apoptosis in Vitro. Tumor cells (0.5 × 106 cells/well) were cultured in 24-well plates in the presence or absence of various concentrations of cannabidiol and/or cannabinoid receptor antagonists for 24 h. Next, the cells were harvested, washed twice in PBS, and analyzed for the induction of apoptosis using the TUNEL or Wright-Giemsa method. To detect apoptosis using the TUNEL method, the cells were washed twice with PBS and fixed with 4% p-formaldehyde for 30 min at room temperature. The cells were next washed with PBS, permeabilized on ice for 2 min, and incubated with fluorescein isothiocyanate-dUTP and TdT (Roche Applied Science, Indianapolis, IN) for 1 h at 37°C and 5% CO2. Five thousand cells were analyzed per sample. To detect apoptosis using the Wright-Giemsa method, centrifuged preparations were stained with Wright-Giemsa stain and observed by light microscopy for signs of apoptosis including nuclear condensation, cell shrinkage, and formation of apoptotic bodies (Jia et al., 2003).
Measurement of Tumor Cell Viability and Induction of Apoptosis in Vivo. Groups of five C57BL/6 mice were i.p. injected with 1 × 106 EL-4 tumor cells suspended in 0.2 ml of PBS. The control mice received PBS alone. Ten days later, the mice were injected with various concentrations of cannabidiol (0, 12.5, or 25 mg/kg) i.p. The mice were sacrificed 24 h later, and the EL-4 tumor cells were harvested from the peritoneal cavity by injecting 5.0 ml of PBS, followed by aspiration of the peritoneal fluid from the cavity. The contaminating red blood cells were removed using red blood lysing solution (Sigma), and the tumor cells were washed twice with PBS. The number of viable cells was determined by trypan blue dye exclusion and apoptosis was determined using the TUNEL assay. The presence of tumor cells in the peritoneal cavity was confirmed by the ability of the cells to grow in vitro and by the phenotype (Thy1+, CD4–, CD8–).
Analysis of Mitochondrial Membrane Potential. Jurkat cells were treated for 16 h with various concentration of CBD. To assess any loss of mitochondrial membrane potential, 3,3′-dihexylcarbocyanine iodide (DiOC6; Invitrogen) was added to the cells for a final concentration of 40 nM, 15 min before the end of the incubation. The cells were then harvested and analyzed using a flow cytometer.
Determination of Reactive Oxygen Species. Jurkat cells were labeled for 1 h with 5 μM carboxy-H2-2′,7′-dichlorofluorescein diacetate (Invitrogen). Next, excess carboxy-H2-2′,7′-dichlorofluorescein diacetate was removed by washing the cells and then suspending them in serum-free, phenol red-free RPMI. The labeled cells were then exposed to various concentrations of CBD for 24 h, and the levels of ROS were determined by flow cytometric analysis and changes in ROS levels were depicted as the percentage increase in fluorescence.
Analysis of Protein Expression by Western Blot Analysis. Protein was isolated from Jurkat cells by freeze thawing and the protein concentration was determined by bicinchoninic acid protein assay kit (Pierce, Rockford, IL). The protein (15-30 μg) was run on a 12% acrylamide gel. The protein was transferred to a nitrocellulose membrane. Nonspecific binding was blocked by incubating the membrane in 5% blotting grade blocker nonfat dry milk (Bio-Rad, Hercules, CA) or bovine serum albumin for 1 h at room temperature. The membrane was rinsed and then washed three times for 10 min with Tris-buffered saline containing 0.1% Tween 20. The membrane was then probed with the specific primary antibody in 5% blocking solution overnight at 4°C. The membrane was then rinsed and washed three times with Tris-buffered saline containing 0.1% Tween 20 and probed with the appropriate horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. The proteins were then visualized after incubation with ECL solution and exposure to X-ray film.
Assessment of Cytosolic Cytochrome c Levels. Jurkat cells were harvested and centrifuged at 600g for 10 min. The cells were then lysed using 100 μl of lysing buffer (75 mM NaCl, 8 mM Na2HPO4,1mMNaH2PO4, 1 mM EDTA, and 350 μg/ml digitonin). Next, the lysates were centrifuged at 12,000g for 5 min. The proteins from resulting fraction (S-100) were quantified, separated by SDS-polyacrylamide gel electrophoresis and probed using monoclonal antibody specific for cytochrome c.

Fig. 1.
Exposure of EL-4 to cannabidiol leads to a reduction in viability and induction of apoptosis in vitro. A, the effect of THC on tumor cell viability was determined by culturing EL-4, in the presence of various concentrations of CBD (1.25, 2.5, 5, or 10 μM) or the vehicle. The viable cell number was determined by trypan blue dye exclusion. B, the effect of CBD on the induction of apoptosis in EL-4 tumor cells was determined by culturing the tumor cells for 24 h in the presence of 2.5, 5, or 10 μM CBD or the vehicle. Apoptosis was quantified using the TUNEL method and the cells were analyzed using a flow cytometer. C, the role of CB1, CB2, and VR1 in mediating the effects of CBD on EL-4 cell viability was determined by culturing EL-4 tumor cells with 5 μM CBD in the presence or absence of CB1 (SR141716A, 5 μM), CB2 (SR144528, 5 μM), or VR1 (CPZ, 5 μM) antagonists. The viable cell number was determined by trypan blue dye exclusion. *, statistically significant difference compared with the vehicle-treated groups, p < 0.05; **, significant difference compared with CBD-treated groups, p < 0.05.
Flow Cytometric Analysis of Phosphorylated Proteins. Tumor cells (0.5 × 106 cells/well) were cultured in 24 wells in the presence of vehicle, cannabidiol (5.0 μM) ± SR144528 (5.0 μM), or cannabidiol ± α-tocopherol (5.0 μM) for 24 h. The cells were harvested and washed twice in PBS. Nonspecific staining was blocked by culturing the cells with Fc Block (BD Pharmingen, San Jose, CA). Next, the cells were fixed for 30 min with 4% paraformaldehyde and then permeabilized by suspending the cells in the BD Perm/Wash solution for 15 min. The presence of the various phosphoproteins was determined by staining with fluorescently labeled monoclonal antibodies specific for phosphorylated and nonphosphorylated ERK, p38, and JNK (Cell Signaling Technology) followed by flow cytometric analysis.
Statistical Analysis. Student’s t test or Tukey Kramer tests were used to compare vehicle and CBD-treated groups. p < 0.05 was considered to be statistically significant.
Results
Exposure of EL-4 to Cannabidiol Leads to a Reduction in Viability and Induction of Apoptosis in Vitro. We examined whether cannabidiol-exposure had an effect on the viability of EL-4, tumor cells in vitro. To this end, the tumor cells were cultured in serum-free medium and exposed to various concentrations of cannabidiol (0, 1.25, 2.5, 5, and 10 μM) for 24 h, and the viability was determined by trypan blue dye exclusion (Fig. 1A). The results showed that exposure to cannabidiol at concentrations of 2.5 μM or greater led to a significant reduction in the number of viable cells. Next, we analyzed the cannabidiol-treated tumor cells for induction of apoptosis by TUNEL staining (Fig. 1B) and demonstrated that cannabidiol induced significant apoptosis in EL-4 tumor cells in vitro. Finally, we examined the effects of CB1-, CB2-, and VR1-selective antagonist on cannabidiol-induced cytotoxicity. EL-4 tumor cells were exposed to cannabidiol (5 μM) in the presence or absence of CB1-, CB2-, or VR1-selective antagonist (SR141716A, SR144528, or CPZ, respectively). EL-4 tumor cells were harvested 24 h later and the cell viability was determined by trypan blue dye exclusion (Fig. 1C). The results showed that treatment with the CB2-selective, but not the CB1- or VR1-selective antagonist was able to significantly inhibit the cannabidiol-induced reduction in cell viability. Together, these results suggested that exposure of EL-4 cells to ≥2.5 μM cannabidiol in vitro led to CB2-dependent cell killing by induction of apoptosis.
Cannabidiol Treatment Leads to Reduced Tumor Burden and Increased Tumor Cell Apoptosis in Vivo.We examined whether treatment of tumor-bearing mice with cannabidiol was effective at killing tumor cells in vivo. To this end, C57BL/6 mice were injected with EL-4 tumor cells (1 × 106). On day 10 of tumor growth, the mice were injected i.p. with various doses of cannabidiol (12.5 or 25 mg/kg) or the vehicle. One day later, the mice were sacrificed and injected with 5 ml of PBS into the peritoneal cavity. The peritoneal fluid was aspirated out and analyzed for viable tumor cells as well as for the presence of apoptotic tumors. The data demonstrated that cannabidiol caused a dose-dependent decrease in the viable tumor cell number found in the peritoneal cavity (Fig. 2A). Furthermore, there was a significant increase in the percentage of apoptotic tumor cells after CBD treatment (Fig. 2B). These data suggested that cannabidiol was effective at killing the EL-4 tumor cells in vivo by inducing apoptosis.

Fig. 2.
Cannabidiol treatment leads to reduced tumor burden and apoptosis in vivo. C57BL/6 mice were injected i.p. on day 0 with 1 × 106 EL-4 tumor cells. On day 10, the mice were treated with various doses of CBD (12.5 or 25 mg/kg i.p.) or the vehicle. One day later, the peritoneal cavity was flushed with 5 ml of PBS, and the tumor cells were collected by aspiration. A, the cell number was determined by trypan blue dye exclusion. B, the tumor cells recovered from the peritoneal cavity were tested for apoptosis using the TUNEL method. The data represent the mean ± S.E.M. from groups of four mice. *, statistically significant difference compared with the vehicle-treated groups, p < 0.05.
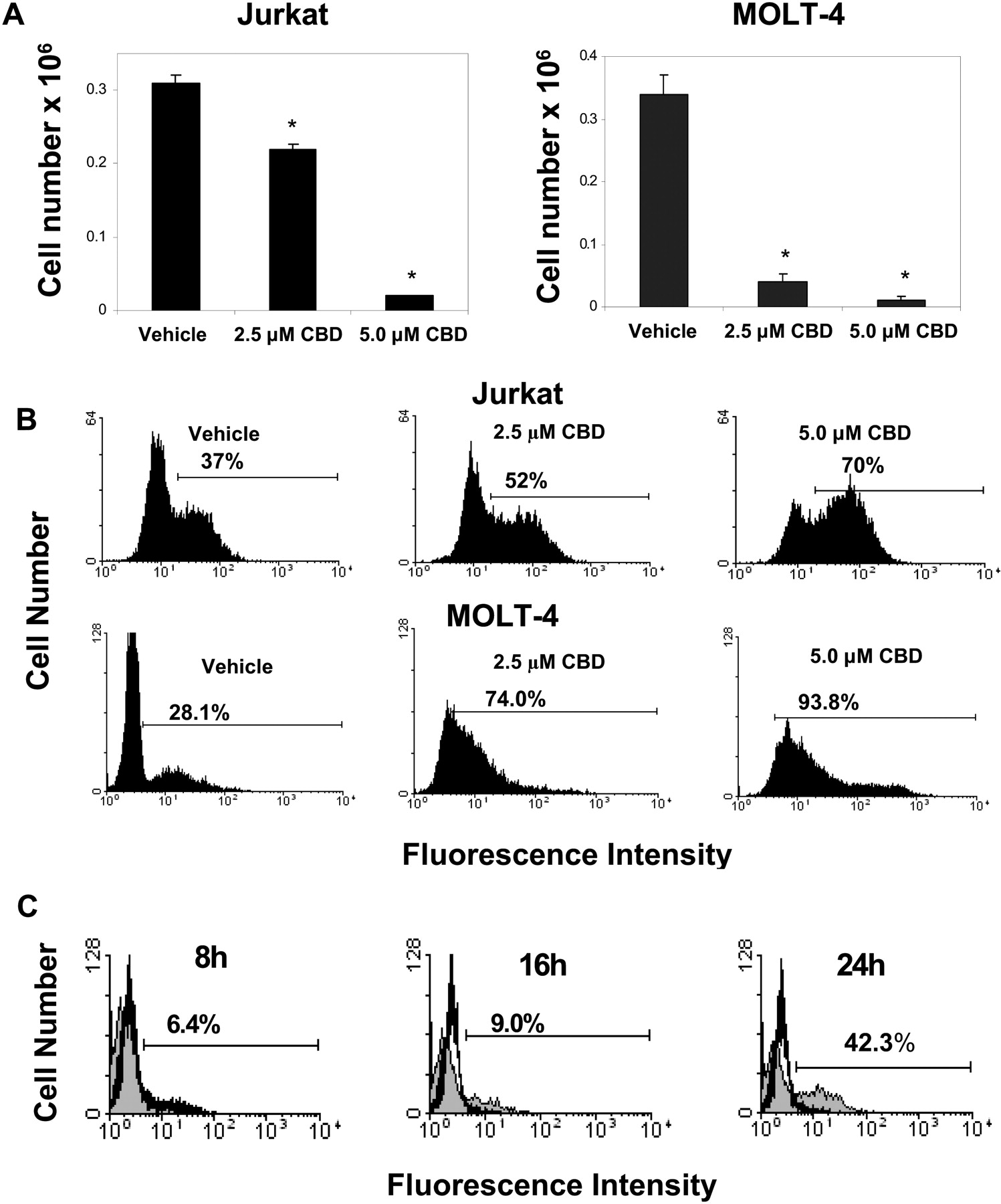
Cannabidiol Induces Apoptosis in Human Leukemia Cells in Vitro. Next, we examined whether exposure of human leukemia cells to cannabidiol would lead to induction of apoptosis. To this end, Jurkat and MOLT-4 cells were exposed to various concentrations of cannabidiol (2.5 and 5 μM) or the vehicle for 24 h, and the effect on cell viability was determined by trypan blue dye exclusion and the induction of apoptosis was determined using the TUNEL method. The results showed that exposure of the Jurkat or MOLT-4 cells to ≥2.5 μM cannabidiol led to significant decrease in the tumor cell viability (Fig. 3A) and an increase in the levels of apoptosis (Fig. 3B). Figure 3B shows a representative experiment using TUNEL assay. Next, experiments were set up to determine the earliest time point in which CBD-induced apoptosis could be detected. To this end, Jurkat cells were cultured with CBD (5.0 μM) for 8, 16, and 24 h (Fig. 3C). The effect of CBD on the induction of apoptosis at the various time points was determined using the TUNEL assay and revealed that significant CBD-induced apoptosis could be detected after 24-h exposure; at the earlier time points, little increase in the percentage of apoptotic cells could be detected. More specifically, after 8 h of exposure to CBD the levels of apoptosis increased from 15.7% (vehicle) to 22.1% (CBD) and after 16 h of exposure to CBD the levels of apoptosis increased from 10.9% (vehicle) to 19.9% (CBD). However, after 24 h of exposure to CBD the levels of apoptosis increased from 6.4% (vehicle) to 48.7% (CBD). Together, these data suggested that cannabidiol exposure can induce apoptosis in human leukemia cells.

Fig. 3.
Cannabidiol induces apoptosis in the Jurkat and MOLT-4 human leukemia cell lines in vitro. Human tumors, Jurkat and Molt-4, in serum free medium, were cultured in the presence of various concentrations of CBD (2.5 and 5 μM), or the vehicle for 24 h. A, the viable cell number was determined by trypan blue dye exclusion. The data represent the mean ± S.E.M. from a representative experiment. B, the induction of apoptosis was determined by TUNEL method. The results from a representative experiment in which human tumor cells cultured with 2.5 or 5 μM CBD or the vehicle is shown. *, statistically significant difference compared with the vehicle-treated groups, p < 0.05. C, Jurkat cells were exposed to CBD (5 μM) for 8, 16, or 24 h. The cells were harvested and analyzed for the induction of apoptosis using the TUNEL assay. The results depicted indicate the increase in the percentage of TUNEL positive cells compared with vehicle-treated cells.
The Role of CB2 in Cannabidiol-Induced Apoptosis. Next, we tested whether cannabidiol was acting through CB1, CB2, or VR1 receptors to induce apoptosis. To this end, Jurkat cells were incubated with 5 μM cannabidiol in the presence of CB1-, CB2-, or VR1-selective antagonists or the vehicle. After 24 h, the viable cell number was determined by trypan blue dye exclusion (Fig. 4A). The results showed that exposure to cannabidiol led to a dramatic reduction in the number of viable tumor cells. However, when the cells were cocultured with the CB2-selective antagonist, the viable cell numbers increased significantly thereby reversing the effect of cannabidiol. In contrast, coculture with either the CB1 or VR1 antagonist was unable to inhibit the effects of cannabidiol on Jurkat viability. Next, we examined whether coculture with the CB2 antagonist would have any effect on cannabidiol-induced apoptosis. To this end, Jurkat cells were incubated with 5 μM cannabidiol in the presence or absence of the CB2-selective antagonist. The presence of apoptotic cells was assayed 24 h later using the TUNEL (Fig. 4B) and Wright-Giemsa staining (Fig. 4C) methods. The results from the TUNEL assay showed that exposure of Jurkat cells to 5.0 μM CBD led to the induction of apoptosis in 83.5% of the cells compared with 3.5% observed in the vehicle-treated cells. However, if the Jurkat cells were pre-exposed to the CB2-selective antagonist, SR144528, the level of CBD-induced apoptosis was significantly reduced to 18.7%. Similar results were seen using the Wright-Giemsa assay, where it was shown that after exposure to 5.0 μM CBD, the majority of the Jurkat cells displayed classic morphological features of apoptosis, including nuclear condensation, cell shrinkage, and formation of apoptotic bodies. In contrast, Jurkat cells pretreated with SR144528 displayed significantly fewer signs of apoptosis after CBD exposure. Treatment with either the CB1 or VR1 antagonist was unable to prevent cannabidiol-induced apoptosis (data not shown). Together, these results suggested that cannabidiol-induced reduction in viable cell number and the induction of apoptosis of Jurkat cells was mediated through CB2.
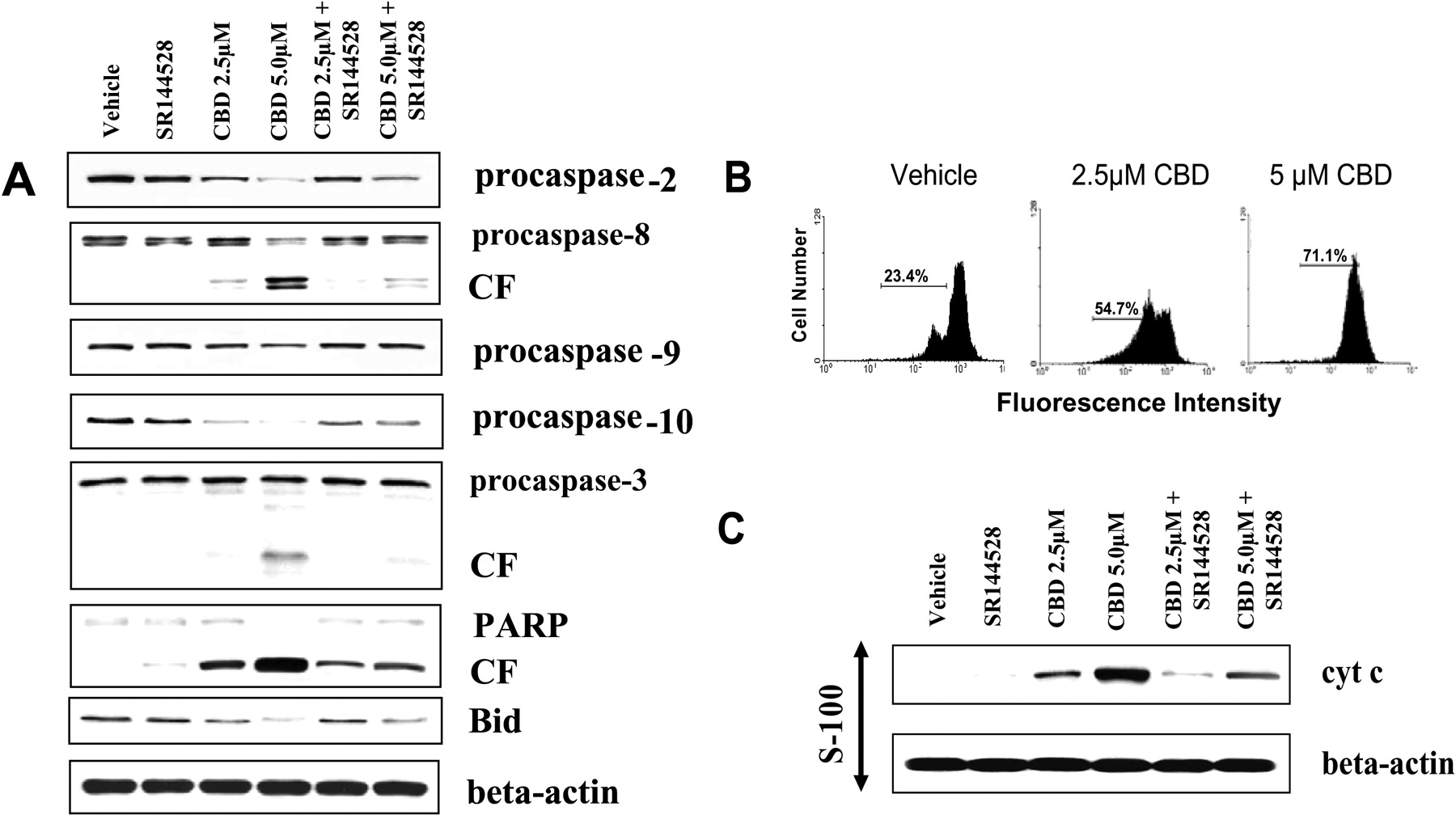
Cannabidiol Induces Activation of the Caspase Cascade through CB2. The activation of the caspase cascade is commonly associated with the induction of apoptosis. Therefore, to further investigate the mechanism of cannabidiol-induced apoptosis, we examined the activation pattern of caspases after CBD exposure. To this end, Jurkat cells were exposed to various concentrations of cannabidiol (2.5 and 5 μM) or the vehicle for 24 h. Next, the cells were harvested and the presence of the various caspases was determined by Western blot analysis (Fig. 5A). The results demonstrate that exposure to CBD at concentrations of 2.5 μM or greater led to activation of the caspase cascade. More specifically, we observed cleavage of caspase-8, and reduction in procaspase-2, -9, and -10, which are thought to be involved in initiating the caspase cascade. In addition, the cleavage of the effector type caspase-3 and poly(ADP-ribose) polymerase were observed after exposure to CBD. Next, the involvement of CB2 was examined by culturing Jurkat cells with the CB2 antagonist SR144528 in the presence or absence of CBD. The results showed that the CB2 antagonist was able to significantly prevent CBD-mediated induction of the caspase cascade. It is noteworthy that CBD-induced cleavage of Bid was observed in Jurkat cells, suggesting possible cross-talk between the intrinsic and extrinsic apoptotic pathways.
CBD Exposure Leads to Loss of Mitochondrial Potential and Release of Cytochrome c. The role of the mitochondria in CBD-induced apoptosis of Jurkat cells was further investigated by examining the effect of CBD on Jurkat mitochondrial membrane potential as well as the levels of cytosolic cytochrome c. Exposure of Jurkat cells to 2.5 μM or greater CBD for 24 h led to a significant reduction in the mitochondrial membrane potential (Fig. 5B). Furthermore, 2.5 μM or greater CBD led to a significant increase in the level of cytosolic cytochrome c (Fig. 5C). Together, these results suggest a direct role of the mitochondria in CBD-induced apoptosis.

Fig. 4.
Cannabidiol-induced apoptosis is mediated through CB2. A, The role of CB1, CB2, and VR1 in mediating the effects of CBD on Jurkat cell viability was determined by culturing Jurkat tumor cells with 5 μM CBD in the presence or absence of the CB1-selective antagonist, (SR141716A, 5 μM), the CB2-selective antagonist, (SR144528, 5 μM), or the VR1-selective antagonist capsazepine (CPZ, 5 μM). The viable cell number was determined by trypan blue dye exclusion and expressed as percentage of control viability. The role of CB2 in mediating CBD-induced apoptosis was determined by culturing Jurkat tumor cells for 24 h with 5 μM CBD in the presence or absence of CB2 antagonist (SR144528, 5 μM). Apoptosis was assayed using the TUNEL (B) and Wright-Giemsa (C) staining methods. *, statistically significant difference compared with the vehicle-treated groups, p < 0.05. **, significant difference compared with CBD-treated groups, p < 0.05.
CBD Induces Increased Generation of ROS. CBD exposure has been associated with increase levels of ROS in glioma cells (Massi et al., 2004). Furthermore, ROS have been associated with activation of the intrinsic apoptotic pathway (Zorov et al., 2000; Singh et al., 2005). Therefore, in the current study, we examined the effect of CBD exposure on Jurkat production of ROS. The results showed that exposure of Jurkat and MOLT-4 cells to 2.5 μM or greater CBD for 24 h led to a significant increase in the levels of ROS production (Fig. 6A). The involvement of the CBD-induced increase in ROS generation in Jurkat apoptosis was further confirmed using the ROS scavengers α-tocopherol and NAC. More specifically, Jurakat cells were exposed to CBD ± α-to-copherol or CBD ± NAC and then analyzed for viable cell number (Fig. 6B), induction of apoptosis (Fig. 6C), and ROS production (Fig. 6D). The results demonstrated that treatment with ROS scavengers led to a significant reduction in CBD-induced Jurkat cell killing and apoptosis. Furthermore, the degree to which the ROS scavengers prevented the effects of CBD on tumor cell killing directly correlated with the ability of the scavengers to reduce the level of CBD-induced ROS.
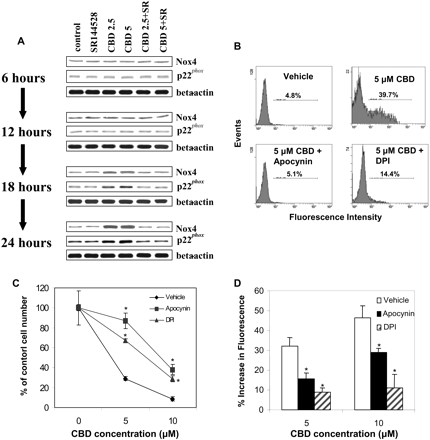
CBD Exposure Leads to Increased Expression of Nox4 and p22phox. NAD(P)H oxidases can play a significant role in the regulation of reactive oxygen species (Suzuki et al., 1998; Lundqvist-Gustafsson and Bengtsson, 1999; Sim et al., 2005). Therefore, we examined whether exposure of Jurkat cells to CBD had an effect on the expression of NAD(P)H oxidases. In initial experiments, the effect of CBD on NAD(P)H oxidases was screened using reverse transcription-polymerase chain reaction. These studies revealed that CBD exposure led to a significant increase in the levels of Nox4 and p22phox, whereas the expression of other NAD(P)H oxidases, including Nox1, Nox2, Nox3, Nox5, p47phox, and p67phox, was unaffected (data not shown). The effect of CBD on Nox4 and p22phox were confirmed by Western blot analysis (Fig. 7A). Furthermore, the role of CB2 in CBD-induced alteration in Nox4 and p22phox expression was confirmed. More specifically, treatment with the CB2-selective antagonist inhibited the CBD-induced effects on Nox4 and p22phox protein expression (Fig. 7A). The role of NAD(P)H oxidases in CBD-induced killing of Jurkat cells was further confirmed using the NAD(P)H oxidase inhibitors DPI and apocynin, demonstrating that treatment with NAD(P)H inhibitors led to a significant reduction in both CBD-induced cell toxicity (Fig. 7B) as well as CBD-induced apoptosis (Fig. 7C) in Jurkat cells. Furthermore, the degree to which the NAD(P)H oxidase inhibitors prevented the effects of CBD on tumor cell killing directly correlated with the ability of the inhibitors to reduce the level of CBD-induced ROS (Fig. 7D).
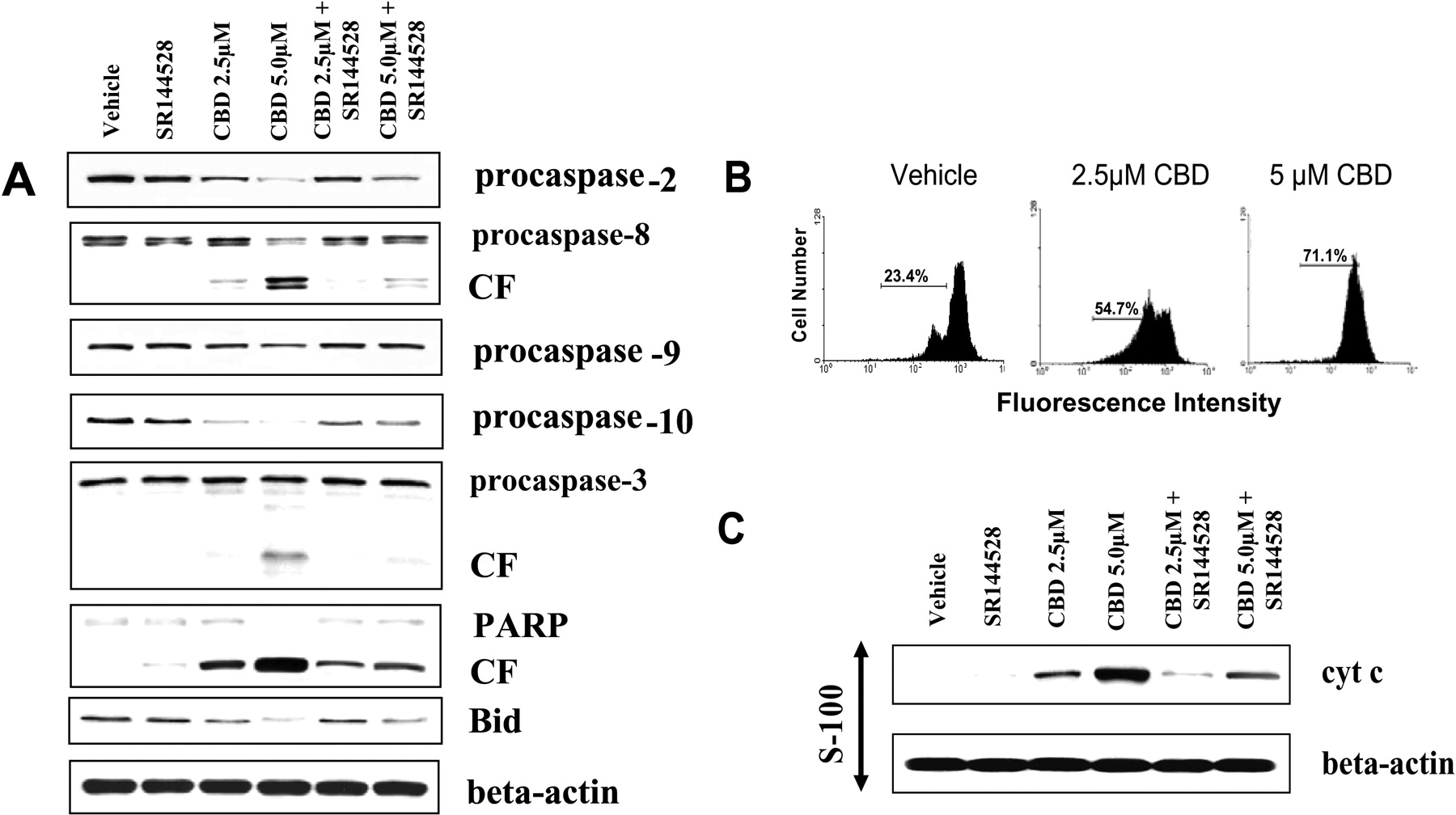
Fig. 5.
Cannabidiol induces activation of the caspase cascade, loss of mitochondrial membrane potential, and release of cytochrome c. A, Jurkat tumor cells were exposed to various concentrations of CBD (2.5 or 5.0 μM) or the vehicle or for 24 h, In addition, the role of CB2 in the CBD-induced changes in caspase activity was monitored by culturing Jurkat cells with CBD (2.5 or 5.0 μM) as well as the CB2-selective antagonist, SR144528. Next, the cells were lysed, the cellular proteins were isolated, and Western analysis was performed. The levels of the procaspases as well as the presence of the cleaved form (CF) of various caspases were examined. B, Jurkat tumor cells were exposed to various concentrations of CBD (2.5 and 5.0 μM) or the vehicle for 24 h. Fifteen minutes before the end of the incubations, DiOC6 was added for a final concentration of 40 nM. The cells were harvested and analyzed by flow cytometry. The percentage of cells with loss of mitochondrial membrane potential is depicted. C, the effect of CBD exposure on the level of cytosolic cytochrome c in Jurkat cells was determined by culturing the cells with CBD (2.5 and 5.0 μM) ± CB2-selective antagonist SR144528 (5.0 μM) for 24 h. The cells were harvested and washed, and cytosolic proteins were analyzed for cytochrome c by Western blot analysis.
The Effect of CBD on the Levels of p-ERK, p-JNK and p-p38 MAPK in Jurkat Cells. The regulation of ERK, JNK, and p38 MAPK have been shown to play an important role in the survival or induction of apoptosis of a number of cell types, including leukemias (Yu et al., 2004; Dasmahapatra et al., 2005). Therefore, we examined whether exposure to CBD had any effect on the levels of the phosphorylated forms of these signaling molecules. To this end, Jurkat cells were exposed for 4 or 16 h to vehicle or 5 μM CBD. Next, the cells were stained with monoclonal antibodies specific for ERK, p-ERK, JNK, p-JNK, p38, and p-p38 MAPK and analyzed by flow cytometric analysis (Fig. 8A). The results demonstrated that exposure to CBD for 16 h led to a significant reduction in the levels of p-p38 MAPK, whereas p-JNK and p-ERK were unaffected. The effect of CBD on the levels of p-p38 were confirmed by Western blot analysis, where it was shown that after 16 h of exposure, CBD led to a significant reduction in the levels of p-p38 (Fig. 8B). No significant effects on p-p38 were seen after 4 or 8 h exposure to CBD. The involvement of CB2 in the down-regulation of these signaling molecules was examined by pre-exposing the Jurkat cells to the CB2-selective antagonist SR144528 and the results showed that the CBD-mediated effects on p-p38 were dependent on CB2 signaling (Fig. 8C). Furthermore, the role of ROS generation in the CBD-mediated reduction of p-p38 was examined by treating Jurkat cells with α-tocopherol before CBD exposure. The results from this experiment demonstrated that treatment with the ROS scavenger was able to prevent the reduction of p-p38 after CBD exposure, suggesting that the effects on ROS generations were upstream of the effects on p38 (Fig. 8C).

Fig. 6.
The effect of CBD on the production of reactive oxygen species in human leukemia cells. A, Jurkat and MOLT-4 tumor cells were exposed to various concentration of CBD (2.5, 5.0, and 10.0 μM) or the vehicle for 24 h and ROS production was determined as described under Materials and Methods and depicted as percentage increase in fluorescence. B, the effect of ROS scavengers on CBD-induced cell toxicity was determined. Jurkat cells were cultured for 24 h with vehicle (control), CBD (5 μM) ± α-tocopherol (20 μM), or CBD (5 μM) ± NAC (10 mM). The viable cell number was determined by trypan blue dye exclusion. The data represent the mean ± S.E.M. of triplicate cultures. C, the effect of α-tocopherol on CBD-induced apoptosis was determined. Jurkat cells were cultured for 24 h with vehicle (control) or CBD (5 μM) ± α-tocopherol (5, 10, or 20 μM). Apoptosis was determined using the TUNEL method. D, the effect of ROS scavengers of CBD-altered ROS levels was determined. Jurkat cells were cultured for 24 h with vehicle (control), CBD (5 μM) ± α-tocopherol (20 μM), or CBD (5 μM) ± NAC (10 mM). ROS production was determined as described under Materials and Methods and depicted as percentage increase in fluorescence. *, statistically significant difference compared with the vehicle-treated groups, p < 0.05.
Discussion
In the current study, we demonstrated that cannabidiol can induce apoptosis in murine as well as human leukemia cells. Cannabidiol-induced apoptosis was mediated through CB2, because treatment with a CB2-selective antagonist, but not a CB1-selective or VR1-selective antagonist, was able to significantly reduce the levels of apoptosis after cannabidiol exposure. Furthermore, cannabidiol was effective in vivo; treatment with cannabidiol led to a significant reduction in tumor burden and an increase in the level of apoptotic tumors in EL-4-bearing mice. Together, these data suggest that cannabidiol may be a novel treatment of leukemia.
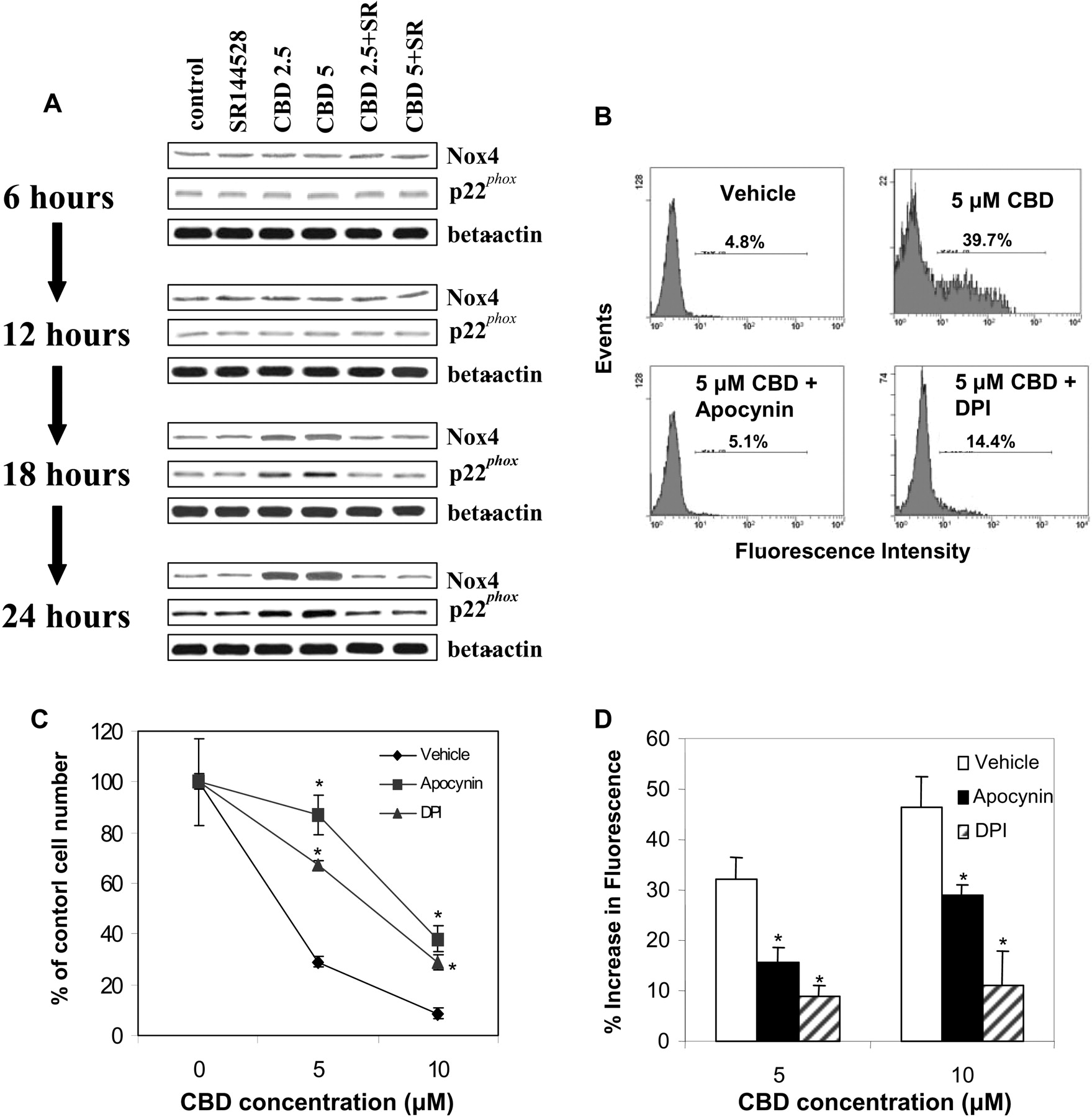
Fig. 7.
CBD exposure leads to increased expression of Nox4 and p22phox. A, Jurkat cells were cultured for various time points with vehicle (control) or CBD (2.5 or 5.0 μM) ± the CB2 selective antagonist SR144528 (5.0 μM). Next, the expression of Nox4 and p22phox in Jurkat cells was determined by Western blot analysis. The effect of DPI and apocynin on CBD-induced toxicity and apoptosis was determined by culturing the cells for 24 h with vehicle (control), CBD (5 μM) ± DPI (5 μM), or CBD (5 μM) ± apocynin (5 μM). The viable cell number was determined by trypan blue dye exclusion (B), and apoptosis was quantified using the TUNEL method (C). D, the effect of NAD(P)H oxidase inhibitors on CBD-altered ROS levels was determined. Jurkat cells were cultured for 24 h with vehicle (control), CBD (5 μM) ± DPI (5 μM), or CBD (5 μM) ± apocynin (5 μM). ROS production was determined as described under Materials and Methods and depicted as percentage increase in fluorescence. *, statistically significant difference compared with the vehicle-treated groups, p < 0.05.
We have demonstrated that treatment with THC leads to significant levels of apoptosis in a number of leukemias (McKallip et al., 2002). Cannabidiol lacks any significant psychotropic activity. Therefore, its use in the treatment of human diseases may be preferable compared with THC or other psychoactive cannabinoids. It is noteworthy that we show that CBD and other cannabinoids such as THC have similar anticancer activity. For example, we demonstrated significant levels of apoptosis in both human and mouse leukemia cell lines after exposure to ≥2.5 μM CBD. These levels were similar to reported levels of THC necessary to induce apoptosis in these cell lines (McKallip et al., 2002). Furthermore, evidence suggests that the doses of CBD used in vitro in the current study were pharmacologically relevant. This is supported by an earlier study in which rats injected with 50 mg/kg of the cannabinoid, THC were shown to exhibit 10 μM THC in the serum within 10 h of administration (Chan et al., 1996).
In a recent study, exposure of glioma cells to 25 μM CBD led to tumor killing by the induction of apoptosis (Massi et al., 2004). In comparison, we found that exposure of leukemia cells to as low as 2.5 μM CBD led to significant levels of apoptosis, suggesting that leukemia cells may be highly sensitive to CBD-induced apoptosis. The current study as well as the report examining the effect of CBD on gliomas (Massi et al., 2004) demonstrated that CB2 is involved in CBD-induced apoptosis, suggesting that the level of CB2 expression may play an important role in the sensitivity of tumor cells to killing by CBD. Therefore, the use of CBD to target CB2 on leukemia cells may prove to be a highly effective and selective strategy for treating this disease.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fig. 8.
The effect of CBD on the expression of p-ERK, p-JNK, and p-p38 in Jurkat cells. A, Jurkat cells were exposed to CBD (5 μM) or vehicle for 4 or 16 h, after which the cells were harvested, and the levels of ERK, p-ERK, JNK, p-JNK, p38, and p-p38 were determined by flow cytometric analysis. B, the effect of CBD exposure (4, 8, and 16 h) on the levels of p38 and p-p38 were analyzed by Western blot analysis. C, the role of CB2 and ROS in CBD-induced alterations in p-p38 levels was examined by treating Jurkat cells with CBD (5 μM), CBD (5 μM) + SR144528 (5.0 μM), or CBD (5 μM) + α-tocopherol (5.0 μM) or vehicle for 16 h, after which the cells were harvested and the levels of p-p38 were determined by flow cytometric analysis.
The role of CB1, CB2, and VR1 in CBD-mediated effects remains controversial. For example, work examining the effects of CBD on glioma cells demonstrated that CB2 was involved in CBD-induced apoptosis (Massi et al., 2004), although the same group of investigators showed that the CBD-mediated suppression of glioma cell migration was through a cannabinoid receptor-independent mechanism (Vaccani et al., 2005). In addition, studies have implicated VR1 in CBD-mediated effects (Bisogno et al., 2001). Furthermore, because of the lipophilic properties of CBD, nonspecific killing through intercalation into the cell membrane may also influence the activity of CBD. However, in the current study, we demonstrated that treatment with the CB2-selective antagonist SR144528 led to significant inhibition of CBD-mediated apoptosis in Jurkat cells. This effect was specific for CB2, in that treatment with CB1- or VR1-antagonists was ineffective at preventing CBD-mediated apoptosis (data not shown).
Compounds that can influence the generation of ROS are currently being examined for their potential use in the treatment of a number of diseases, including cancer (Kang et al., 2004; Kim et al., 2004; Chang et al., 2005; Hu and Brindle, 2005; Lebedeva et al., 2005). However, little has been reported regarding the ability of cannabinoids to regulate the generation of ROS. In a study examining the effect of CBD on glioma cells, CBD-induced apoptosis was related to increased ROS generation (Massi et al., 2004). In the current study, we demonstrated a similar role of ROS in CBD-induced apoptosis in Jurkat cells. More specifically, we demonstrated that exposure to CBD led to an increase in ROS production and that treatment with ROS scavengers significantly inhibited CBD-induced apoptosis in Jurkat cells. It is noteworthy that the level by which the ROS scavengers inhibited CBD-induced apoptosis and loss of cell viability correlated with the level of inhibition of CBD-induced increase in ROS levels, further supporting the role of ROS in CBD-induced apoptosis. Together, these data suggest that regulation of ROS generation by cannabidiol may be an effective strategy to treat malignancies of the immune system.
NAD(P)H oxidases are a group of enzymes involved in the regulation of ROS production (Zorov et al., 2000; Singh et al., 2005). Nox4 and p22phox are NAD(P)H oxidases that play a role in a number of processes, including cell survival and apoptosis (Pedruzzi et al., 2004; Vaquero et al., 2004; Martyn et al., 2006). However, the specific effects of these enzymes in these processes remain unclear. For example, studies examining pancreatic cancer cell survival suggested that growth factor-induced ROS generation controlled by various NAD(P)H oxidases, including Nox4, promoted tumor cell survival (Vaquero et al., 2004; Edderkaoui et al., 2005). In contrast, it was reported that Nox4 played a significant role in mediating apoptosis in human aortic smooth muscle cells after exposure to 7-ketocholesterol (Pedruzzi et al., 2004). So far, expression of these enzymes and their contribution to the induction of apoptosis in leukemia cells has not been reported. In the current study, we demonstrated that CBD exposure led to increased expression of the NAD(P)H oxidases Nox4 and p22phox, suggesting the possibility that regulation of these enzymes and the subsequent generation of ROS may play a significant role in the induction of apoptosis in leukemia cells. Therefore, targeting Nox4 and p22phox using CBD may be a novel approach for treating malignancies of the immune system.
The mechanism by which CBD exposure leads to increased expression of Nox4 and p22phox is not clear. However, initial observations in this study showing that pretreatment with a CB2-selective antagonist, but not a CB1- or VR1-selective antagonist, significantly inhibited the CBD-mediated increase in Nox4 and p22phox expression, suggests that CBD regulation of these enzymes is mediated through CB2 activation. It is noteworthy that the mechanism of action seems to be distinct from that of THC, which can lead to increased ceramide production and has been associated with G-protein-coupled receptor signaling (Galve-Roperh et al., 2000; Rueda et al., 2000). In the current study, we were unable to block CBD-induced apoptosis using various inhibitors of ceramide generation or pertussis toxin, which blocks G-protein coupled receptor signaling (data not shown). Taken together, these data suggest that although both THC and CBD exposure can lead to the induction of apoptosis in leukemia cells, their mechanisms of action may be distinct from one another. Therefore, treatments using the combination of THC and CBD may significantly enhance the efficacy of tumor killing. To date, this approach has not been reported for the treatment of leukemia or lymphoma. However, initial studies demonstrated that the combination of CBD and THC was better tolerated than THC alone (reviewed in Russo, 2006), possibly allowing for the use of increased levels of THC without the unwanted psychoactive side effects. In fact, it was shown that CBD could antagonize some of the undesirable effects of THC, such as its psychoactive properties, by blocking conversion of THC to the more psychoactive 11-hydroxy-THC (Bornheim and Grillo, 1998). On the other hand, as shown in the current study and by other investigators examining the effects on gliomas (Massi et al., 2004), CBD alone has been shown to possess potent antitumor activity. Taken together, these studies suggest that the combination of THC and CBD may produce a powerful combination for the treatment of various cancers, including gliomas and malignancies of the immune system.
In addition, results from this study demonstrated that CBD exposure led to the reduction of p-p38 levels, whereas p-ERK and p-JNK levels were unaffected. Support for the role of p38 in CBD-induced apoptosis comes from reports demonstrating that CBD exposure led to reduced levels of p-p38 in PC-12 neuronal cells (Esposito et al., 2006). Furthermore, in a separate study, it was shown that reduction in p-p38 levels in Jurkat cells led to the induction of apoptosis (Nemoto et al., 1998). Taken together, these results suggest that CBD-induced apoptosis is regulated, at least in part, through alterations in p38 signaling.
In summary, the current study demonstrates that CBD-induced apoptosis may constitute a novel approach to treat malignancies of the immune system. CBD has been reported to be nonpsychoactive, making it more attractive than its psychoactive counterpart, THC, as a therapeutic agent. Here we found that the CBD-induced apoptosis was dependent on CB2 receptor signaling. CB2 is expressed almost exclusively on immune cells. Therefore, use of CB2 receptor agonists such as CBD may be significantly less toxic to nonimmune cells. Furthermore, we demonstrated, for the first time, that exposure of leukemia cells to CBD led to increased production of ROS which was mediated through regulation of Nox4 and p22phox, suggesting a novel role of CBD in the regulation of ROS production in leukemia cells. Thus, further research on the use of CBD to target transformed immune cells could lead to a new and highly selective anticancer agent.
Footnotes
-
This work was supported in part by grants from National Institutes of Health (R01-DA016545, R21-DA014885, K12-DA14041, and P50-DA05274), The American Cancer Society (IRG-100036) and The Jeffress Memorial Trust Fund (J-741).
-
ABBREVIATIONS: THC, Δ9-tetrahydrocannabinol; CBD, cannabidiol; ROS, reactive oxygen species; PBS, phosphate-buffered saline; SR141716A; CB1, cannabinoid receptor 1; CB2, cannabinoid receptor 2; DPI, diphenylene iodinium; CPZ, capsazepine; VR1, vanilloid receptor 1; NAC, N-acetylcysteine; TUNEL, terminal deoxynucleotidyl transferase dUTP nick-end labeling; DiOC6, 3,3′-dihexylcarbocyanine iodide; ERK, extracellular signal-regulated kinase; JNK, c-Jun NH2-terminal kinase; MAPK, mitogen-activated protein kinase.
-
- Received February 28, 2006.
- Accepted June 5, 2006.
- The American Society for Pharmacology and Experimental Therapeutics