| |||

| |||
| Names | |||
|---|---|---|---|
| IUPAC name
Azinous acid
| |||
| Preferred IUPAC name
Hydroxylamine (only preselected[1]) | |||
Other names
| |||
| Identifiers | |||
3D model (JSmol)
|
|||
| 3DMet | |||
| ChEBI | |||
| ChEMBL | |||
| ChemSpider | |||
| ECHA InfoCard | 100.029.327 | ||
| EC Number |
| ||
| 478 | |||
| KEGG | |||
| MeSH | Hydroxylamine | ||
PubChem CID
|
|||
| RTECS number |
| ||
| UNII | |||
CompTox Dashboard (EPA)
|
|||
| |||
| |||
| Properties | |||
| NH2OH | |||
| Molar mass | 33.030 g·mol−1 | ||
| Appearance | Vivid white, opaque crystals | ||
| Density | 1.21 g cm−3 (at 20 °C)[2] | ||
| Melting point | 33 °C (91 °F; 306 K) | ||
| Boiling point | 58 °C (136 °F; 331 K) /22 mm Hg (decomposes) | ||
| Soluble | |||
| log P | −0.758 | ||
| Acidity (pKa) | 6.03 ([NH3OH]+) | ||
| Basicity (pKb) | 7.97 | ||
| Structure | |||
| Tricoordinated at N, dicoordinated at O | |||
| Trigonal pyramidal at N, bent at O | |||
| 0.67553 D | |||
| Thermochemistry | |||
Heat capacity (C)
|
46.47 J/(K·mol) | ||
Std molar
entropy (S⦵298) |
236.18 J/(K·mol) | ||
Std enthalpy of
formation (ΔfH⦵298) |
−39.9 kJ/mol | ||
| Hazards | |||
| GHS labelling: | |||
    
| |||
| Danger | |||
| H200, H290, H302, H312, H315, H317, H318, H335, H351, H373, H400 | |||
| P201, P202, P234, P260, P261, P264, P270, P271, P272, P273, P280, P281, P301+P312, P302+P352, P304+P340, P305+P351+P338, P308+P313, P310, P312, P314, P321, P322, P330, P332+P313, P333+P313, P362, P363, P372, P373, P380, P390, P391, P401, P403+P233, P404, P405, P501 | |||
| NFPA 704 (fire diamond) | |||
| Flash point | 129 °C (264 °F; 402 K) | ||
| 265 °C (509 °F; 538 K) | |||
| Lethal dose or concentration (LD, LC): | |||
LD50 (median dose)
|
408 mg/kg (oral, mouse); 59–70 mg/kg (intraperitoneal mouse, rat); 29 mg/kg (subcutaneous, rat)[3] | ||
| Safety data sheet (SDS) | ICSC 0661 | ||
| Related compounds | |||
Related hydroxylammonium salts
|
|||
Related compounds
|
|||
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa).
| |||



Hydroxylamine (also known as hydroxyammonia) is an inorganic compound with the chemical formula NH2OH. The compound is in a form of a white hygroscopic crystals.[4] Hydroxylamine is almost always provided and used as an aqueous solution. It is consumed almost exclusively to produce Nylon-6. The oxidation of NH3 to hydroxylamine is a step in biological nitrification.[5]
History[edit]
Hydroxylamine was first prepared as hydroxylammonium chloride in 1865 by the German chemist Wilhelm Clemens Lossen (1838-1906); he reacted tin and hydrochloric acid in the presence of ethyl nitrate.[6] It was first prepared in pure form in 1891 by the Dutch chemist Lobry de Bruyn and by the French chemist Léon Maurice Crismer (1858-1944).[7][8] The coordination complex ZnCl2(NH2OH)2 (zinc dichloride di(hydroxylamine)), known as Crismer's salt, releases hydroxylamine upon heating.[9]
Production[edit]
Hydroxylamine or its salts (salts containing hydroxylammonium cations [NH3OH]+) can be produced via several routes but only two are commercially viable. It is also produced naturally as discussed in a section on biochemistry.
From nitric oxide[edit]
NH2OH is mainly produced as its sulfuric acid salt, hydroxylammonium hydrogen sulfate ([NH3OH]+[HSO4]−), by the hydrogenation of nitric oxide over platinum catalysts in the presence of sulfuric acid.[10]
Raschig process[edit]
Another route to NH2OH is the Raschig process: aqueous ammonium nitrite is reduced by HSO−3 and SO2 at 0 °C to yield a hydroxylamido-N,N-disulfonate anion:
This anion is then hydrolyzed to give hydroxylammonium sulfate [NH3OH]2SO4:
- N(OH)(SO−3)2 + H2O → NH(OH)(SO−3) + HSO−4
- 2 NH(OH)(SO−3) + 2 H2O → [NH3OH]2SO4 + SO2−4
Solid NH2OH can be collected by treatment with liquid ammonia. Ammonium sulfate, [NH4]2SO4, a side-product insoluble in liquid ammonia, is removed by filtration; the liquid ammonia is evaporated to give the desired product.[4] The net reaction is:
A base then frees the hydroxylamine from the salt:
- [NH3OH]Cl + NaOCH2CH2CH2CH3 → NH2OH + NaCl + CH3CH2CH2CH2OH[4]
Other methods[edit]
Julius Tafel discovered that hydroxylamine hydrochloride or sulfate salts can be produced by electrolytic reduction of nitric acid with HCl or H2SO4 respectively:[11][12]
- HNO3 + 3 H2 → NH2OH + 2 H2O
Hydroxylamine can also be produced by the reduction of nitrous acid or potassium nitrite with bisulfite:
- HNO2 + 2 HSO−3 → N(OH)(OSO−2)2 + H2O → NH(OH)(OSO−2) + HSO−4
- NH(OH)(OSO−2) + [H3O]+ → [NH3OH]+ + HSO−4 (100 °C, 1 h)
Hydrochloric acid disproportionates nitromethane to hydroxylamine hydrochloride and carbon monoxide via the hydroxamic acid.[citation needed]
A direct production of hydroxylamine from molecular nitrogen is also possible in water plasma.[13]
Reactions[edit]
Hydroxylamine reacts with electrophiles, such as alkylating agents, which can attach to either the oxygen or the nitrogen atoms:
- R−X + NH2OH → R−O−NH2 + HX
- R−X + NH2OH → R−NH−OH + HX
The reaction of NH2OH with an aldehyde or ketone produces an oxime.
This reaction is useful in the purification of ketones and aldehydes: if hydroxylamine is added to an aldehyde or ketone in solution, an oxime forms, which generally precipitates from solution; heating the precipitate with an inorganic acid then restores the original aldehyde or ketone.[14]
Oximes such as dimethylglyoxime are also employed as ligands.
NH2OH reacts with chlorosulfonic acid to give hydroxylamine-O-sulfonic acid:[15]
- HO−S(=O)2−Cl + NH2OH → NH2−O−S(=O)2−OH + HCl
When heated, hydroxylamine explodes. A detonator can easily explode aqueous solutions concentrated above 80% by weight, and even 50% solution might prove detonable if tested in bulk.[16][17] In air, the combustion is rapid and complete:
- 4 NH2OH + O2 → 2 N2 + 6 H2O
Absent air, pure hydroxylamine requires stronger heating and the detonation does not complete combustion:
- 3 NH2OH → N2 + NH3 + 3 H2O
Partial isomerisation to the amine oxide H3N+−O− contributes to the high reactivity.[18]
Conjugate oxoacids[edit]
Hydroxylamine yields via oxidation to azinic acid.[citation needed] Further attempted oxidation yields nitric acid, and finally orthonitrate.
More hydroxyl radicals substitute the hydrogen atoms to yield azonous acid & azorous acid.
Functional group[edit]

Substituted derivatives of hydroxylamine are known. When the hydroxyl or an amine hydrogen is substituted, such a molecule is called (respectively) an O- or N-hydroxylamine. In general N-hydroxylamines are more common. Examples are N-tert-butylhydroxylamine or the glycosidic bond in calicheamicin. N,O-Dimethylhydroxylamine is a precursor to Weinreb amides.
Similarly to amines, one can distinguish hydroxylamines by their degree of substitution: primary, secondary and tertiary. When stored exposed to air for weeks, secondary hydroxylamines degrade to nitrones.[19]
N-organylhydroxylamines, R−NH−OH, where R is an organyl group, can be reduced to amines R−NH2:[20]
- R−NH−OH (Zn, HCl) → R−NH2 + ZnO
Synthesis[edit]
Amine oxidation with benzoyl peroxide is the most common method to synthesize hydroxylamines. Care must be taken to prevent over-oxidation to a nitrone. Other methods include:
- Hydrogenation of an oxime
- Alkylating a precursor hydroxylamine
- Amine oxide pyrolysis (the Cope reaction)
Uses[edit]
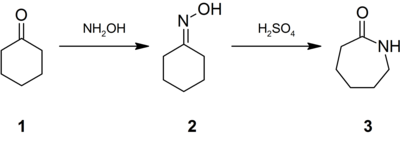
Conversion of cyclohexanone to caprolactam involving the Beckmann rearrangement.

Approximately 95% of hydroxylamine is used in the synthesis of cyclohexanone oxime, a precursor to Nylon 6.[10] The treatment of this oxime with acid induces the Beckmann rearrangement to give caprolactam (3).[21] The latter can then undergo a ring-opening polymerization to yield Nylon 6.[22]
Laboratory uses[edit]
Hydroxylamine and its salts are commonly used as reducing agents in myriad organic and inorganic reactions. They can also act as antioxidants for fatty acids.
High concentrations of hydroxylamine are used by biologists to introduce mutations by acting as a DNA nucleobase amine-hydroxylating agent.[23] In is thought to mainly act via hydroxylation of cytidine to hydroxyaminocytidine, which is misread as thymidine, thereby inducing C:G to T:A transition mutations.[24] But high concentrations or over-reaction of hydroxylamine in vitro are seemingly able to modify other regions of the DNA & lead to other types of mutations.[24] This may be due to the ability of hydroxylamine to undergo uncontrolled free radical chemistry in the presence of trace metals and oxygen, in fact in the absence of its free radical affects Ernst Freese noted hydroxylamine was unable to induce reversion mutations of its C:G to T:A transition effect & even considered hydroxylamine to be the most specific mutagen known.[25] Practically, it has been largely surpassed by more potent mutagens such as EMS, ENU, or nitrosoguanidine, but being a very small mutagenic compound with high specificity, it found some specialized uses such as mutation of DNA packed within bacteriophage capsids,[26] & mutation of purified DNA in vitro.[27]

This route also involves the Beckmann Rearrangement, like the conversion from cyclohexanone to caprolactam.

An alternative industrial synthesis of paracetamol developed by Hoechst–Celanese involves the conversion of ketone to a ketoxime with hydroxylamine.
Some non-chemical uses include removal of hair from animal hides and photographic developing solutions.[2] In the semiconductor industry, hydroxylamine is often a component in the "resist stripper", which removes photoresist after lithography.
Hydroxylamine can also be used to better characterize the nature of a post-translational modification onto proteins. For example, poly(ADP-Ribose) chains are sensitive to hydroxylamine when attached to glutamic or aspartic acids but not sensitive when attached to serines.[28] Similarly, Ubiquitin molecules bound to serines or threonines residues are sensitive to hydroxylamine, but those bound to lysine (isopeptide bond) are resistant.[29]
Biochemistry[edit]
In biological nitrification, the oxidation of NH3 to hydroxylamine is mediated by the ammonia monooxygenase (AMO).[5] Hydroxylamine oxidoreductase (HAO) further oxidizes hydroxylamine to nitrite.[30]
Cytochrome P460, an enzyme found in the ammonia-oxidizing bacteria Nitrosomonas europea, can convert hydroxylamine to nitrous oxide, a potent greenhouse gas.[31]
Hydroxylamine can also be used to highly selectively cleave asparaginyl-glycine peptide bonds in peptides and proteins.[32] It also bonds to and permanently disables (poisons) heme-containing enzymes. It is used as an irreversible inhibitor of the oxygen-evolving complex of photosynthesis on account of its similar structure to water.
Safety and environmental concerns[edit]
With a theoretical decomposition energy of about 5 kJ/g, hydroxylamine is an explosive, and aqueous solutions above 80% can be easily detonated by detonator or strong heating under confinement.[16] [17] At least two factories dealing in hydroxylamine have been destroyed since 1999 with loss of life.[33] It is known, however, that ferrous and ferric iron salts accelerate the decomposition of 50% NH2OH solutions.[34] Hydroxylamine and its derivatives are more safely handled in the form of salts.
It is an irritant to the respiratory tract, skin, eyes, and other mucous membranes. It may be absorbed through the skin, is harmful if swallowed, and is a possible mutagen.[35]
See also[edit]
References[edit]
- ^ "Front Matter". Nomenclature of Organic Chemistry : IUPAC Recommendations and Preferred Names 2013 (Blue Book). Cambridge: The Royal Society of Chemistry. 2014. p. 993. doi:10.1039/9781849733069-FP001. ISBN 978-0-85404-182-4.
- ^ a b Lide, David R., ed. (2006). CRC Handbook of Chemistry and Physics (87th ed.). Boca Raton, FL: CRC Press. ISBN 0-8493-0487-3.
- ^ Martel, B.; Cassidy, K. (2004). Chemical Risk Analysis: A Practical Handbook. Butterworth–Heinemann. p. 362. ISBN 978-1-903996-65-2.
- ^ a b c Greenwood and Earnshaw. Chemistry of the Elements. 2nd Edition. Reed Educational and Professional Publishing Ltd. pp. 431–432. 1997.
- ^ a b Lawton, Thomas J.; Ham, Jungwha; Sun, Tianlin; Rosenzweig, Amy C. (2014-09-01). "Structural conservation of the B subunit in the ammonia monooxygenase/particulate methane monooxygenase superfamily". Proteins: Structure, Function, and Bioinformatics. 82 (9): 2263–2267. doi:10.1002/prot.24535. ISSN 1097-0134. PMC 4133332. PMID 24523098.
- ^ W. C. Lossen (1865) "Ueber das Hydroxylamine" (On hydroxylamine), Zeitschrift für Chemie, 8 : 551-553. From p. 551: "Ich schlage vor, dieselbe Hydroxylamin oder Oxyammoniak zu nennen." (I propose to call it hydroxylamine or oxyammonia.)
- ^ C. A. Lobry de Bruyn (1891) "Sur l'hydroxylamine libre" (On free hydroxylamine), Recueil des travaux chimiques des Pays-Bas, 10 : 100-112.
- ^ L. Crismer (1891) "Préparation de l'hydroxylamine cristallisée" (Preparation of crystalized hydroxylamine), Bulletin de la Société chimique de Paris, series 3, 6 : 793-795.
- ^ Walker, John E.; Howell, David M. (1967). "Dichlorobis(hydroxylamine)zinc(II) (Crismer's Salt)". Inorganic Syntheses. Vol. 9. pp. 2–3. doi:10.1002/9780470132401.ch2. ISBN 9780470132401.
- ^ a b Ritz, Josef; Fuchs, Hugo; Perryman, Howard G. (2000). "Hydroxylamine". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a13_527. ISBN 978-3527306732.
- ^ James Hale, Arthur (1919). The Manufacture of Chemicals by Electrolysis (1st ed.). New York: D. Van Nostrand Co. p. 32. Retrieved 5 June 2014.
manufacture of chemicals by electrolysis hydroxylamine 32.
- ^ Osswald, Philipp; Geisler, Walter (1941). Process of preparing hydroxylamine hydrochloride (US2242477) (PDF). U.S. Patent Office.
- ^ Zhang, Xiaoping; Su, Rui; Li, Jingling; Huang, Liping; Yang, Wenwen; Chingin, Konstantin; Balabin, Roman; Wang, Jingjing; Zhang, Xinglei; Zhu, Weifeng; Huang, Keke; Feng, Shouhua; Chen, Huanwen (2024). "Efficient catalyst-free N2 fixation by water radical cations under ambient conditions". Nature Communications. 15 (1) 1535. doi:10.1038/s41467-024-45832-9. PMC 10879522. PMID 38378822.
- ^ Ralph Lloyd Shriner, Reynold C. Fuson, and Daniel Y. Curtin, The Systematic Identification of Organic Compounds: A Laboratory Manual, 5th ed. (New York: Wiley, 1964), chapter 6.
- ^ Wiberg, Egon; Wiberg, Nils (2001). Inorganic Chemistry. Academic Press. pp. 675–677. ISBN 978-0-12-352651-9.
- ^ a b Iwata, Yusaku; Koseki, Hiroshi; Hosoya, Fumio (2003-01-01). "Study on decomposition of hydroxylamine/water solution". Journal of Loss Prevention in the Process Industries. 16 (1): 41–53. doi:10.1016/S0950-4230(02)00072-4. ISSN 0950-4230.
- ^ a b Bretherick's Handbook of Reactive Chemical Hazards. ISBN 9780081009710. Retrieved 2023-08-28.
- ^ Kirby, AJ; Davies, JE; Fox, DJ; Hodgson, DR; Goeta, AE; Lima, MF; Priebe, JP; Santaballa, JA; Nome, F (28 February 2010). "Ammonia oxide makes up some 20% of an aqueous solution of hydroxylamine". Chemical Communications. 46 (8): 1302–4. doi:10.1039/b923742a. PMID 20449284.
- ^ Hamer, Jan; Macaluso, Anthony. "Nitrones". Chemical Reviews: 476. doi:10.1021/cr60230a006.
- ^ Smith, Michael and Jerry March. March's advanced organic chemistry : reactions, mechanisms, and structure. New York. Wiley. p. 1554. 2001.
- ^ Clayden, Jonathan; Greeves, Nick; Warren, Stuart (2012). Organic chemistry (2nd ed.). Oxford University Press. p. 958. ISBN 978-0-19-927029-3.
- ^ Nuyken, Oskar; Pask, Stephen (25 April 2013). "Ring-Opening Polymerization—An Introductory Review". Polymers. 5 (2): 361–403. doi:10.3390/polym5020361.
- ^ Waugh, Robbie; Leader, David J.; McCallum, Nicola; Caldwell, David (2006). "Harvesting the potential of induced biological diversity". Trends in Plant Science. 11 (2). Elsevier BV: 71–79. doi:10.1016/j.tplants.2005.12.007. ISSN 1360-1385. PMID 16406304.
- ^ a b Busby, Stephen; Irani, Meher; de Crombrugghe, Benoít (1982). "Isolation of mutant promoters in the Escherichia coli galactose operon using local mutagenesis on cloned DNA fragments". Journal of Molecular Biology. 154 (2). Elsevier BV: 197–209. doi:10.1016/0022-2836(82)90060-2. ISSN 0022-2836. PMID 7042980.
- ^ Hollaender, Alexander (1971). Chemical Mutagens : Principles and Methods for Their Detection Volume 1. Boston, MA: Springer US. p. 41. ISBN 978-1-4615-8968-6. OCLC 851813793.
- ^ Hong, J.-S.; Ames, B. N. (1971-12-01). "Localized Mutagenesis of Any Specific Small Region of the Bacterial Chromosome". Proceedings of the National Academy of Sciences. 68 (12): 3158–3162. Bibcode:1971PNAS...68.3158H. doi:10.1073/pnas.68.12.3158. ISSN 0027-8424. PMC 389612. PMID 4943557.
- ^ Forsberg, Susan. "Hydroxylamine Mutagenesis of plasmid DNA". PombeNet. University of Southern California. Retrieved 9 December 2021.
- ^ Langelier, Marie-France; Billur, Ramya; Sverzhinsky, Aleksandr; Black, Ben E.; Pascal, John M. (2021-11-18). "HPF1 dynamically controls the PARP1/2 balance between initiating and elongating ADP-ribose modifications". Nature Communications. 12 (1): 6675. Bibcode:2021NatCo..12.6675L. doi:10.1038/s41467-021-27043-8. ISSN 2041-1723. PMC 8602370. PMID 34795260.
- ^ Kelsall, Ian R.; Zhang, Jiazhen; Knebel, Axel; Arthur, J. Simon C.; Cohen, Philip (2019-07-02). "The E3 ligase HOIL-1 catalyses ester bond formation between ubiquitin and components of the Myddosome in mammalian cells". Proceedings of the National Academy of Sciences. 116 (27): 13293–13298. Bibcode:2019PNAS..11613293K. doi:10.1073/pnas.1905873116. ISSN 0027-8424. PMC 6613137. PMID 31209050.
- ^ Arciero, David M.; Hooper, Alan B.; Cai, Mengli; Timkovich, Russell (1993-09-01). "Evidence for the structure of the active site heme P460 in hydroxylamine oxidoreductase of Nitrosomonas". Biochemistry. 32 (36): 9370–9378. doi:10.1021/bi00087a016. ISSN 0006-2960. PMID 8369308.
- ^ Caranto, Jonathan D.; Vilbert, Avery C.; Lancaster, Kyle M. (2016-12-20). "Nitrosomonas europaea cytochrome P460 is a direct link between nitrification and nitrous oxide emission". Proceedings of the National Academy of Sciences. 113 (51): 14704–14709. Bibcode:2016PNAS..11314704C. doi:10.1073/pnas.1611051113. ISSN 0027-8424. PMC 5187719. PMID 27856762.
- ^ Bornstein, Paul; Balian, Gary (1977). Cleavage at Asn-Gly bonds with Hydroxylamine. Methods in Enzymology. Vol. 47(Enzyme Struct., Part E). pp. 132–45. doi:10.1016/0076-6879(77)47016-2. PMID 927171.
{{cite book}}: CS1 maint: multiple names: authors list (link) - ^ Japan Science and Technology Agency Failure Knowledge Database Archived 2007-12-20 at the Wayback Machine.
- ^ Cisneros, L. O.; Rogers, W. J.; Mannan, M. S.; Li, X.; Koseki, H. (2003). "Effect of Iron Ion in the Thermal Decomposition of 50 mass% Hydroxylamine/Water Solutions". J. Chem. Eng. Data. 48 (5): 1164–1169. doi:10.1021/je030121p.
- ^ MSDS Sigma-Aldrich
Further reading[edit]
- Hydroxylamine[permanent dead link]
- Walters, Michael A. and Andrew B. Hoem. "Hydroxylamine." e-Encyclopedia of Reagents for Organic Synthesis. 2001.
- Schupf Computational Chemistry Lab
- M. W. Rathke A. A. Millard "Boranes in Functionalization of Olefins to Amines: 3-Pinanamine" Organic Syntheses, Coll. Vol. 6, p. 943; Vol. 58, p. 32. (preparation of hydroxylamine-O-sulfonic acid).